

Computational Biology and Chemistry
Physical Geometric Algorithms
Introduction
Some of the most challenging and influential opportunities for Physical Geometric Algorithms (PGA) arise in developing and applying information technology to understand the molecular machinery of the cell. Our recent work (e.g., [1-20]) shows that many PGA techniques may be fruitfully applied to the challenges of computational molecular biology. PGA research may lead to computer systems and algorithms that are useful in structural molecular biology, proteomics, and rational drug design.

Concomitantly, a wealth of interesting computational problems arise in proposed methods for discovering new pharmaceuticals. I'll briefly discuss some recent results from my lab, including new algorithms for interpreting X-ray crystallography [14, 17, 16] and NMR (nuclear magnetic resonance) data [3,9,6,19,10,5,7,18,4], disease classification using mass spectrometry of human serum [12], and protein redesign [13]. Our algorithms have recently been used, respectively, to reveal the enzymatic architecture of organisms high on the CDC bioterrorism watch-list [17,16], for probabilistic cancer classification from human peripheral blood [12], and to redesign an antibiotic-producing enzyme to adenylate a novel substrate [13]. I'll overview these projects, and highlight some of the algorithmic and computational challenges.
Our papers [Search Medline, DBLP, CCSB, PDF Reprints]
1. Background and Significance
In the post-genomic era, key problems in molecular biology center on the determination and exploitation of three-dimensional protein structure and function. For example, modern drug design techniques use protein structure to understand how a drug can bind to an enzyme and inhibit its function. Structural proteomics will require high-throughput experimental techniques, coupled with sophisticated computer algorithms for data analysis and experiment planning.

Amy Anderson (our wonderful collaborator at the University of Connecticut), Ryan Lilen, Robert O'Neil, and Bruce Donald, along with Robert Stroud from the University of California at San Francisco, have revealed the architecture of a parasite's enzyme, which could help researchers reduce the threat of certain diseases among those with weak immune systems. Part of the enzyme's structure is depicted behind the team and in O'Neil's hands. (Read more...)
My laboratory develops novel computational methods to enable high-throughput structural and functional studies of proteins. A key focus is structural genomics, whose goal is (in the broadest terms) to determine the three-dimensional structures of all proteins in nature, through a combination of direct experiments and theoretical analysis. Proteins are the worker molecules in every living thing. By determining the structures of proteins, we are better able to understand how each protein functions normally and how faulty protein structures can cause disease. Scientists can use the structures of disease-related proteins to help develop new medicines and diagnostic techniques.
PGA research in computational structural biology and proteomics can assist in our long-range goal of understanding biopolymer interactions in systems of significant biochemical as well as pharmacological interest. At the molecular level, many genes provide the blueprint for proteins, and it remains very expensive and time-consuming to determine what these proteins do, and how they do it. This page reviews some novel algorithms to build three-dimensional models of proteins to better understand protein mechanism and function.
Our work spans a number of projects in computational structural biology, including algorithms for automated assignment and structure determination in NMR structural biology [3,9,6,19,10,5,7,18,4], protein redesign [13], computer-aided drug design [1,15,13], computational molecular replacement in X-ray crystallography [14,17,16], and other related projects [2, 11,8]. To pursue research in the field of structural genomics is to study the geometric structures of proteins. Structural genomics is a field born of the the marriage between computer science and biology. The goal is to develop new technology - specifically, computer algorithms - to enable the determination of 10,000 new protein structures in 10 years. This would have an enormous impact on our understanding of disease mechanisms, and our ability to target drugs to specific protein targets. Some examples include:

Targeting the molecular architecture of infectious diseases...
Modern automated techniques are revolutionizing many aspects of
biology, for example, supporting extremely fast gene sequencing and
massively-parallel gene expression testing. Protein structure
determination, however, remains a long, hard, and expensive
task. High-throughput, automated, algorithmic methods are required in
order to apply modern techniques such as computer-aided drug design on
a much larger scale. For example, to analyze non-crystallographic
symmetry in X-ray diffraction data of biopolymers, one must
"recognize" a finite subgroup of SO(3) (the Lie group of 3D rotations) out of a
large set of molecular orientations. The problem may be reduced to
clustering in SO(3) modulo a finite group, and solved efficiently by
"factoring" into a clustering on the unit circle followed by
clustering on the 2-sphere S², plus some
group-theoretic calculations [CRANS, 14].
This yields a polynomial-time algorithm that is efficient in practice,
and which enabled us to collaborate with biological crystallographers
to reveal the architecture of a parasite's enzyme, which could help
researchers reduce the threat of certain diseases among those with
weak immune systems [17,16].
...By Exploiting Symmetry.
[CRANS, 14] is a new algorithm, helpful in
solving the structures of certain symmetric biological macromolecules
by X-ray crystallography. As pointed out by Isupov and Lebedev
(Acta. Cryst. D, 2007):
- "Presence of noncrystallographic symmetry (NCS) can be
advantageous for structure determination by molecular replacement
(MR). In many cases the orders and directions of NCS axes can be
determined on the basis of experimental data using the self-rotation
function (SRF; Rossmann & Blow, 1962). Translational NCS can be
detected using the native Patterson synthesis. A comparison of
experimental functions and functions generated from MR solutions is a
good validation tool. Two of many examples are given by Makino et
al. (2005) and Keillor et al. (2003).
The presence of NCS (excluding that generated by pure translation) allows phase improvement by averaging (Bricogne, 1974). Often, a conserved oligomer is a better search model for MR than a monomer. In many cases, the oligomer models are readily available in the PDB and these are routinely used for MR trials in MR pipelines (Keegan & Winn, 2007; Long et al., 2008). The locked rotation function (LRF) and locked translation function (LTF) were developed to build an oligomer from monomers and to orient it in accordance with the SRF (Tong, 2001). The resulting oligomer is used in a conventional translation-function (TF) search. NCS analysis has to be performed with special care when NCS is used directly for structure determination (as in the LRF/LTF method). An interesting example of misleading the SRF was reported by Asojo et al. (2003).
The point-group symmetry of an oligomer is an approximate symmetry and its deviation from the exact symmetry may be too large for methods based on averaging of the Patterson function (LRF/LRF) to be successful. The selection of cross-rotation function (cross RF) peaks obeying NCS
[CRANS, 14] is free of this disadvantage."Cancer biology, geometry, and proteomics. We employed geometric techniques from statistical estimation and machine learning to develop an algorithm for cancer proteomics, in which we use data from a mass spectrometer to distinguish between healthy and diseased blood in humans [12]. Geometry pervades our work: [12] can be viewed as an investigation of the geometry of the oncoproteome (the space of cancer proteins) as projected onto the mass-to-charge ratios of their proteolytic digest. Nuclear vector replacement for automated NMR resonance assignments [6,10] is essentially a matching problem on a quotient space of orientations, induced by a quadratic form ξ on S². ξ is parameterized by SO(3) , and 3D structural homology detection from unassigned NMR data (enabling rapid fold determination) can be performed by combinatorial optimization, searching over SO(3) to minimize a functional that compares distributions generated by ξ's image of the bond vectors from putative database protein models [9,10,5,7].
Combatting Leukemia: drug design and protein-protein interactions. Working with a team of collaborators, we used a new algorithm for predicting the free energy of protein:ligand interactions to develop novel allosteric small-molecule inhibitors of the binding of two leukemia-associated proteins. While numerous inhibitors of enzymatic activity had previously been developed, the development of inhibitors of protein-protein interactions has only recently come to the forefront as a viable approach. Allosteric inhibition of such protein-protein interactions presents a number of advantages, including not having to compete for binding with the partner protein. However, there are very few previous examples of such inhibitors. Our inhibitors block the binding of RUNX1 to CBFβ, two proteins whose translocations play a critical role in the development of acute myeloid leukemia and acute lymphocytic leukemia.The results are reported in the cover article of Chemistry & Biology (Cell Press, 2007). Treatment of human leukemia cell lines with our inhibitor results in changes in morphology indicative of increased differentiation versus untreated cells, providing support for this as a potential therapeutic approach.

In each of our research projects, computational techniques are central, and the applications present intriguing problems to computer scientists who design algorithms and implement systems. For example, the techniques we introduced for automated NMR resonance assignment [6,10,3,9,7] and protein structure determination [19,18,5,7] are an instance a general approach to combinatorial problem solving in which constraints on the solution are enforced in an order determined by the strength of the evidence for them. This approach, which has analogies to the Celera whole-genome shotgun sequencing algorithm, also presents a flock of fascinating questions from the point of view of theoretical computer science (cf. Richard Karp's Keynote address, Computational Systems Bioinformatics Conference, 2003).
2. Results in NMR Structural Genomics
In this section, I attempt to illustrate the general themes introduced above, by way of specific examples and results. Here is an article in the popular press about this work. In the following, the NMR biophysics requires some technical description; some readers may prefer to skip to the higher-level discussion on Protein Fold Determination (2b) or even to Section 3.

At Duke University our lab uses the 800, 600, and 500 mHz NMR spectrometers located in our building. The largest is a state-of-the-art ultra-highfield 800 MHz Varian Inova NMR spectrometer fully configured with four channels and a 1H, 13C, 15N triple probe with three axis gradients, with a triple-resonance cold-probe. We also have access to the 950 mHz spectrometer in Kannapolis (shown here).
2a. Nuclear Vector Replacement for Automated NMR Resonance Assignment and Structure Determination
In X-ray crystallography, the molecular replacement technique (used in [14,17,16]) allows solution of the crystallographic phase problem when a "close'' or homologous structural model is known a priori, thereby facilitating rapid structure determination. In contrast, a key bottleneck in NMR structural biology is the resonance assignment problem. One would hope that knowing a structural model ahead of time could expedite assignments. We recently reported an automated procedure for high-throughput NMR resonance assignment for a protein of known structure, or of an homologous structure [6]. Our algorithm performs Nuclear Vector Replacement (NVR) by Expectation/Maximization (EM) to compute assignments. NVR correlates experimentally-measured NH residual dipolar couplings (RDCs) and chemical shifts to a given a priori whole-protein 3D structural model. The algorithm requires only uniform 15N -labeling of the protein, and processes unassigned HN-15 N HSQC spectra, HN-15N RDCs, and sparse HN-HN NOE's (dNNs), all of which can be acquired in a fraction of the time needed to record the traditional suite of experiments used to perform resonance assignments. NVR runs in minutes and efficiently assigns the (HN, 15N) backbone resonances as well as the sparse dNNs N-NOESY spectrum, in O(n3) time. We tested NVR on NMR data from 3 proteins using 20 different alternative structures, all determined either by X-ray crystallography or by different NMR experiments (without RDCs). When NVR was run on NMR data from the 76-residue protein, human ubiquitin (matched to four structures, including one mutant/homolog), we achieved 100% assignment accuracy. Similarly good results were obtained in experiments with the 56-residue streptococcal protein G (99%) and the 129-residue hen lysozyme (100%) when they were matched by NVR to 16 3D structural models. Our success in assigning 1UD7, a mutant of ubiquitin, suggests that NVR could be applied more broadly to assign spectra based on homologous structures. Thus, NVR could play a role in structural genomics.
2b. Protein Fold Determination via Unassigned Residual Dipolar Couplings
In 2002, we extended NVR to a second application, 3D structural homology detection, and demonstrated that NVR is able to identify structural homologies between remote amino acid sequences from a database of structural models. Our first paper on protein fold determination using unassigned residual dipolar couplings was received in September, 2002 and published in RECOMB in April, 2003 [9]. Our other papers on this topic include [5 (August, 2003)] and [10,7]. One goal of the structural genomics initiative is the identification of new protein folds. Sequence-based structural homology prediction methods are an important means for prioritizing unknown proteins for structure determination. However, an important challenge remains: two highly dissimilar sequences can have similar folds -- how can we detect this rapidly, in the context of structural genomics? High-throughput NMR experiments, coupled with novel algorithms for data analysis, can address this challenge. We reported an automated procedure for detecting 3D structural homologies from sparse, unassigned protein NMR data. Our method identifies the 3D structural models in a protein structural database whose geometries best fit the unassigned experimental NMR data. It does not use sequence information and is thus not limited by sequence homology. The method can also be used to confirm or refute structural predictions made by other techniques such as protein threading or sequence homology. The method requires only uniform 15N-labeling of the protein and processes unassigned HN-15N residual dipolar couplings, which can be acquired in a couple of hours. Our experiments on NMR data from 5 different proteins demonstrate that the method identifies closely related protein folds, despite low-sequence homology between the target protein and the computed model.
2c. Novel NMR Structure Determination Algorithms

Bruce Donald, Chris Langmead, and Lincong Wang have developed new algorithms that interpret NMR data to reveal a protein's shape and molecular architecture. NMR surveys a protein's molecular structure, and, conceptually, can be thought of as using tiny, spectroscopic protractors and rulers to generate a network of geometric measurements (Article, J. Biomol. NMR 29(2), J. Biomol. NMR 29(3)).
Three recent papers [19,20,18] make contributions to the method of determining protein structures by solution NMR spectroscopy using residual dipolar couplings (RDCs) as the main restraints. These contributions, I believe, may be valuable not only to the NMR community in particular and structural genomics in general, but also to structural biologists more broadly. This is because in both experimental and computational structural biology, exact computational methods have been, for the most part, elusive to date. Second, rigorous comparisons of structures derived from NMR vs. X-ray crystallography are made possible by our techniques, and these comparisons should be of general interest.
In contrast to (e.g.) simulated annealing approaches, our algorithm is combinatorially-precise [18], and is built upon the exact solutions for computing backbone (φ,ψ) angles from RDC data and systematic search. [19] is the first NMR structure determination algorithm that simultaneously uses exact solutions, systematic search and only 2 RDCs per residue. (A systematic search is a search over all possible conformations (solutions) that employs a provable pruning strategy that guarantees pruned conformations need not be considered further). Our first contribution is the derivation of low-degree polynomial equations for computing, exactly and in constant time, dihedral (φ,ψ) angles from RDCs measured on a single internuclear vector v in two different aligning media. The easily computable exact solutions eliminate the need for one-dimensional grid-search previously employed to compute the directions of v or two-dimensional grid-search to compute (φ,ψ) angles. Furthermore, these equations are very general and can easily be extended to compute both the backbone and sidechain dihedral angles from RDC data measured on any single vector in two aligning media. And, our method can be applied mutatis mutandis to derive similar equations for computing dihedral angles from RDCs in nucleic acids. Compared with other algorithms for computing backbone structures using RDCs, our algorithm achieves similar accuracies but requires less data, relies less on statistics from the PDB and does not depend on molecular dynamics. Since RDCs can be acquired and assigned much more quickly than NOEs in general, our results show it is possible to compute structures very rapidly and inexpensively using mainly RDC restraints.
3. Analytic vs. Synthetic

Redesigning the PheA domain of Gramicidin Synthetase leads to a new understanding of the enzyme's mechanism and selectivity. Biochemistry (2006).
Most of the work described above concentrates on algorithms and system for analyzing biological data and biological problems. However, the techniques we develop can also be applied to synthetic problems such as a protein engineering. For example, in collaboration with Prof. Amy Anderson, we recently developed a novel ensemble-based scoring and search algorithm for protein redesign, and applied it to modify the substrate specificity of an antibiotic-producing enzyme in the non-ribosomal peptide synthetase (NRPS) pathway [13]. Realization of novel molecular function requires the ability to alter molecular complex formation. Enzymatic function can be altered by changing enzyme-substrate interactions via modification of an enzyme's active site. A redesigned enzyme may either perform a novel reaction on its native substrates or its native reaction on novel substrates. A number of computational approaches have been developed to address the combinatorial nature of the protein redesign problem. These approaches typically search for the global minimum energy conformation among an exponential number of protein conformations. We developed a novel algorithm for protein redesign, which combines a statistical mechanics-derived ensemble-based approach to computing the binding constant with the speed and completeness of a branch-and-bound pruning algorithm. In addition, we developed an efficient deterministic approximation algorithm, capable of approximating our scoring function to arbitrary precision. Our algorithm is the first provable ε-approximation algorithm for estimating partition functions for protein flexibility.
In practice, our approximation algorithm decreases the execution time of the mutation search by a factor of ten. To test our method, we examined the Phe-specific adenylation domain of the NRPS gramicidin synthetase A (GrsA-PheA). We used ensemble scoring, via a rotameric approximation to the partition functions of the bound and unbound states for GrsA-PheA, to predict binding of the wildtype protein and a previously described mutant (selective for leucine), and second, to switch the enzyme specificity toward leucine, using two novel active site sequences computationally predicted by searching through the space of possible active site mutations. The top scoring in silico mutants were created in the wetlab and activity and binding were experimentally measured. These tested mutations exhibit the desired change in specificity from Phe to Leu. We also predicted and tested mutants that created enzyme activity (virtually non-existent in the wildtype) for 4 charged amino acid substrates. Our ensemble-based algorithm which flexibly models both protein and ligand using rotamer-based partition functions, has application in enzyme redesign, the prediction of protein-ligand binding, and computer-aided drug design.
In PNAS (2009), we report a computational structure-based redesign of the phenylalanine adenylation domain of the non-ribosomal peptide synthetase enzyme Gramicidin S Synthetase A (GrsA-PheA) for a set of non-cognate substrates for which the wildtype enzyme has little or virtually no specifcity. Experimental validation of a set of top-ranked computationally-predicted enzyme mutants shows signifcant improvement in the specifcity for the target substrates. We further present enhancements to the methodology for computational enzyme redesign that are experimentally shown to result in signifcant additional improvements in the target substrate specifcity. The mutant with the highest activity for a non-cognate substrate exhibits 1/6 of the wildtype enzyme/wildtype substrate activity, further confrming the feasibility of our computational approach. Our results suggest that structure-based protein design can identify active mutants different from those selected by evolution.
This result represents a computational approach to reprogramming enzyme specificity, with the ultimate goal of combinatorial biosynthesis for small-molecule diversity. We are studying a family of enzymes responsible for the biosynthesis of hundreds of pharmaceutically-active peptide-like products. Understanding these enzyme functions will elucidate how natural biological products (e.g., antibiotics) are synthesized in vivo. To modify enzyme function, we are developing computational techniques to plan structurally-based site-directed mutations. By re-engineering the active site(s) to operate on different substrates, we hope to modify these different enzymatic steps with an eye to potential reprogramming of those steps for combinatorial biosynthesis. This opens the door to the possibility of using our redesigned enzymes for in vivo combinatorial chemistry, to create candidate drug leads for new antibiotics and other drugs. We are applying our algorithms to NRPS modules, whose products include natural antibiotics, antifungals, antivirals, immunosuppressants, and siderophores. NRPS have multiple domains with individual functions acting in an assembly-line fashion. We are modifying the active sites to switch the specificity of the amino acid-accepting domains from their natural substrates, to different amino acids. The modifications are planned and analyzed in silico, by developing new algorithms based on techniques from geometric algorithms, robotics, machine vision, and scientific computation. Our "enzyme reprogramming" could allow the modified NRPS to synthesize different modified peptides. Exploration of the combinatorial space of new NRPS "programs" will generate a large number of new compounds, which could then be screened for pharmaceutical activity.
4. Outlook

One conference we like is RECOMB. Here is Bruce Donald, right, at RECOMB'04 (with Herbert Edelsbrunner).
There is much to be done. A primary focus will be to explore novel computational methods in structural biology, specifically, new algorithms for NMR resonance assignments and protein structure determination, with applications to structural genomics. I am particularly interested in new algorithms for structural biology using only a minimal number of inexpensive, fast experiments. I'm also interested in collaborating with structural biologists to develop novel algorithms and apply them to biological systems of significant biochemical and pharmacological importance. A model for this kind of work is our collaboration on the structure of dihydrofolate reductase-thymidylate synthase (DHFR-TS) from Cryptosporidium hominis [14,17,16]. Cryptosporidium is an organism high on the bioterrorism list of the Center for Disease Control (CDC), a Category B bioterrorist threat. Agents/diseases that fall under this category are given the second-highest priority, because they are moderately easy to disseminate; result in moderate morbidity rates and low mortality rates; and require specific enhancements of CDC's diagnostic and treatment capacities. There is currently no drug therapy for cryptosporidosis. The enzyme DHFR-TS is in the sole de novo biosynthetic pathway for the pyrimidine deoxyribonucleotide dTMP, and therefore an attractive drug target. Solving the structure of DHFR-TS from C. hominis opens the door to species-specific drug design, exploiting both structural and biophysical differences between the human enzyme and Cryptosporidium DHFR-TS.
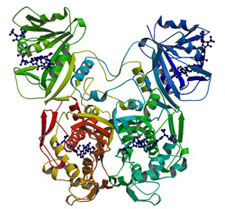
Structure of Dihydrofolate Reductase-Thymidylate Synthase from Cryptosporidium hominis. In J. Biol. Chem. 2003.
We collaborated with Dr. Amy Anderson's lab to determine the structure of DHFR-TS from C. hominis, revealing a unique linker domain containing an 11-residue alpha helix that has extensive interactions with the opposite DHFR-TS monomer of the homodimeric enzyme [17,16]. Analysis of the structure of DHFR-TS from C. hominis and of previously solved structures of DHFR-TS from Plasmodium falciparum (a.k.a. malaria) and Leishmania major reveals that the linker domain primarily controls the relative orientation of the DHFR and TS domains. Using the tertiary structure of the linker domains, we have been able to place a number of protozoa in two distinct and dissimilar structural families corresponding to two evolutionary families and provide the first structural evidence validating the use of DHFR-TS as a tool of phylogenetic classification.
I am also interested more broadly in proteomics and functional genomics, both in developing novel algorithms for proteomic problems and bringing to bear structural, geometric, and biophysical insights (and algorithms) for proteomics. For example, in collaboration with the Norris-Cotton Cancer Center at Dartmouth, we are exploring oncoproteomic target selection using mass spectrometry [12]. We developed an algorithm called Q5 for probabilistic classification of healthy vs. disease whole serum samples using mass spectrometry. Q5 is the first closed-form, exact solution to the problem of classification of complete mass spectra of a complex protein mixture. It employs a discriminant back-projection algorithm to compute clues as to the molecular identities of differentially-expressed proteins and peptides. Q5 analyzes whole spectrum Surface-Enhanced Laser Desorption/Ionization Time of Flight (SELDI-TOF) Mass Spectrometry (MS) data, and was demonstrated on four real datasets from complete, complex SELDI spectra of human blood serum. We achieved sensitivity, specificity, and positive predictive values above 97% on three ovarian cancer datasets and one prostate cancer dataset. The Q5 method outperforms previous full-spectrum complex sample spectral classification techniques, and represents the first attempt to compute the molecular identities of the differentially-expressed proteins in two important MS data sets for ovarian and prostate cancer. Further investigation of our lead proteins and peptide fragments may enhance our understanding of the molecular basis of oncogenisis and could potentially lead to new therapeutic targets.
Hany Farid (left), Bruce Donald (seated), and Ryan Lilien have developed an algorithm named Q5 that uses data from a mass spectrometer to distinguish between healthy and diseased blood (Details).
As discussed above (Sec. 3), we are designing and implementing planning algorithms to propose site-directed mutations for protein redesign. We are developing a general planner that can reprogram the specificity of many NRPS domains, from many biological systems. The results of our algorithms are being compared to known crystal structures, to biochemical activity assays, and to crystal structures of the modified domains bound to the proposed substrates. We are developing a predictive model for when and how well our planner will work, by characterizing the complexity, correctness, and completeness of our algorithms. We believe these algorithms may be generally useful to the structural biology community, for studies of protein-ligand binding and protein redesign. Our provable approximation algorithm represents a new technique for computer-assisted drug design, and a novel approach for docking flexible ligands to flexible active sites [13]. In the future, we will extend and apply our algorithms to the redesign of other enzymes, including polyketide synthase (PKS) systems, which synthesize polyketide products such as erythromycin, rapamycin and tetracycline. I believe there are broad potential applications of our techniques for modeling protein flexibility, redesigning enzymes, and evaluating the biophysical processes of binding and catalysis in protein biochemistry, and that these goals can (hopefully) be realized after a lot more hard work in developing geometric algorithms, provably-good approximation algorithms, statistical methods, and an array of algorithmic techniques for handling noise and uncertainty in combinatorial geometry and computational biophysics.
Selected Bibliography
A more complete list of publications can be found here.
-
C. Bailey-Kellogg, J. J. Kelley III, R. Lilien, and B. R. Donald.
Physical geometric algorithms for structural molecular biology.
In the Special Session on Computational Biology & Chemistry, Proceedings IEEE Int'l Conf. on Robotics and Automation (ICRA-2001), pages 940-947, May 2001. -
C. Bailey-Kellogg, J. J. Kelley III, C. Stein, and B. R. Donald.
Reducing mass degeneracy in SAR by MS by stable isotopic labeling.
Jour. Comp. Biol., 8(1):19-36, 2001. -
C. Bailey-Kellogg, A. Widge, J. J. Kelley III, M. J. Berardi, J. H.
Bushweller, and B. R. Donald.
The NOESY Jigsaw: Automated protein secondary structure and main-chain assignment from sparse, unassigned NMR data.
Jour. Comp. Biol., 3-4(7):537-558, 2000. -
C. Langmead and B. R. Donald.
Extracting structural information using time-frequency analysis of protein NMR data.
In Proceedings of The Fifth Annual International Conference on Research in Computational Molecular Biology (RECOMB), pages 164-175. ACM Press, April 2001. -
C. Langmead and B. R. Donald.
3D structural homology detection via unassigned residual dipolar couplings.
In Proceedings of the IEEE Computer Society Bioinformatics Conference (CSB), pages 209-217, Stanford, August 2003. -
C. Langmead and B. R. Donald.
An expectation/maximization nuclear vector replacement algorithm for automated NMR resonance assignments.
Jour. Biomolecular NMR, 29(2):111-138, 2004. -
C. Langmead and B. R. Donald.
High-throughput 3D structural homology detection via NMR resonance assignment.
In Proceedings of the IEEE Computational Systems Bioinformatics Conference (CSB), Stanford, CA, August 2004 pp. 278-289. -
C. Langmead, C. R. McClung, and B. R. Donald.
A maximum entropy algorithm for rhythmic analysis of genome-wide expression patterns.
In Proceedings of the IEEE Computer Society Bioinformatics Conference (IEEE CSB), pages 237-245, August 2002. -
C. Langmead, A. Yan, R. Lilien, L. Wang, and B. R. Donald.
A polynomial-time nuclear vector replacement algorithm for automated NMR resonance assignments.
In Proceedings of The Seventh Annual International Conference on Research in Computational Molecular Biology (RECOMB), pages 176-187, Berlin, Germany, April 2003. ACM Press. -
C. Langmead, A. Yan, R. Lilien, L. Wang, and B. R. Donald.
A polynomial-time nuclear vector replacement algorithm for automated NMR resonance assignments.
Jour. Comp. Biol., 11(2-3):277-298, 2004. -
C. Langmead, A. Yan, C. R. McClung, and B. R. Donald.
Phase-independent rhythmic analysis of genome-wide expression patterns.
Journal of Computational Biology, 10(3-4):521-536, 2003. -
R. Lilien, H. Farid, and B. R. Donald.
Probabilistic disease classification of expression-dependent proteomic data from mass spectrometry of human serum.
Journal of Computational Biology, 10(6):925-946, 2003. - C. Chen, I. Georgiev, A. Anderson, and B. R. Donald. Computational Structure-Based Redesign of Enzyme Activity. Proc. Nat. Acad. Sci. U.S.A. (PNAS) (2009).
-
Ryan H. Lilien, Chris Bailey-Kellogg, Amy C. Anderson, and Bruce R. Donald.
A subgroup algorithm to identify cross-rotation peaks consistent with non-crystallographic symmetry.
Acta Crystallographica Section D, 60(6):1057-1067, Jun 2004. -
Ryan H. Lilien, Mohini Sridharan, and Bruce R. Donald.
Identification of Novel Small Molecule Inhibitors of Core-Binding Factor Dimerization by Computational Screening against NMR Molecular Ensembles.
Technical Report TR2004-492, Dartmouth College, Computer Science, Hanover, NH, March 2004. -
R. O'Neil, R. Lilien, B. R. Donald, R. Stroud, and A. Anderson.
The crystal structure of dihydrofolate reductase-thymidylate synthase from Cryptosporidium hominis reveals a novel architecture for the bifunctional enzyme.
Jour. Eukaryotic Microbiology, 50(6):555-556, 2003. -
R. O'Neil, R. Lilien, B. R. Donald, R. Stroud, and A. Anderson.
Phylogenetic classification of protozoa based on the structure of the linker domain in the bifunctional enzyme, dihydrofolate reductase-thymidylate synthase.
Jour. Biol. Chem., 278(52):52980-52987, 2003. -
L. Wang and B. R. Donald.
Analysis of a systematic search-based algorithm for determining protein backbone structure from a minimal number of residual dipolar couplings.
In Proceedings of the IEEE Computational Systems Bioinformatics Conference (CSB), Stanford, CA, August 2004 pp. 319-330. -
L. Wang and B. R. Donald.
Exact solutions for internuclear vectors and backbone dihedral angles from NH residual dipolar couplings in two media, and their application in a systematic search algorithm for determining protein backbone structure.
Jour. Biomolecular NMR, 29(3):223-242, 2004. -
L. Wang, R. Mettu, R. Lilien, and B. R. Donald.
An exact algorithm for determining protein backbone structure from NH residual dipolar couplings.
In Proceedings of the IEEE Computer Society Bioinformatics Conference (CSB), pages 611-612, Stanford, August 2003.