

Chittaranjan Tripathy

My research interests lie in Computational Structural Biology, Bioinformatics, and Applied Algorithms.
My current research focuses on developing efficient and robust algorithms for (i) Protein Structure Determination from a minimal amount of experimental data from Nuclear Magnetic Resonance (NMR) spectroscopy, and (ii) the Protein Loop Modeling Problem. My research interests also include Protein Design, and the study of protein dynamics and Intrinsically Disordered Proteins (IDPs), with an emphasis on sparse-data algorithms that use the knowledge mined from the big data involving protein structures, functions and experimental measurements, available to the scientific community. My research draws on tools and techniques from the biophysics and biochemistry of proteins, algebraic geometry, combinatorial optimization, machine learning and robotics.
Protein structure determination from sparse NMR data is a challenging task. Most structure determination protocols currently require a large amount of experimental data, and use stochastic conformational sampling methods to compute protein structures, and therefore, lack any algorithmic guarantee (e.g. completeness, and optimility) on the quality of the solution or running time. In contrast, my research has demonstrated the feasibility of determining protein structure from sparse NMR measurements using polynomial-time deterministic algorithms which can potentially save both time and money, and accelerate the structure determination process.
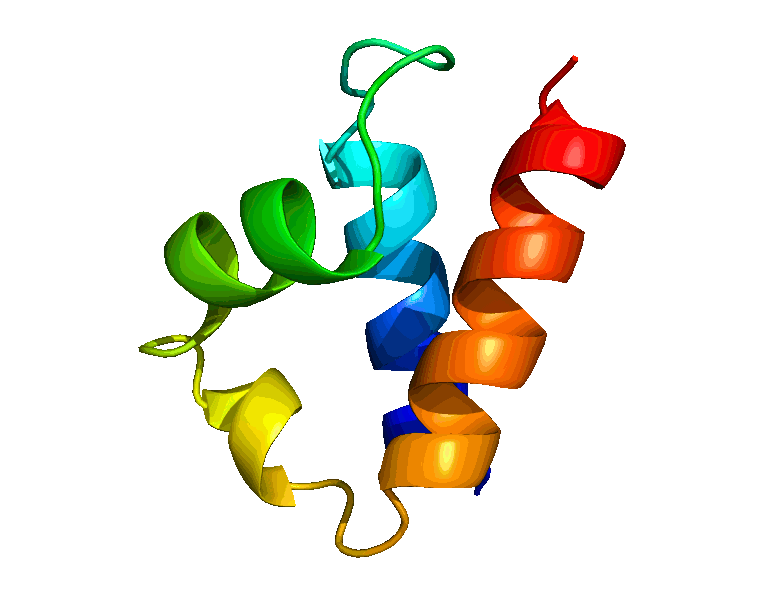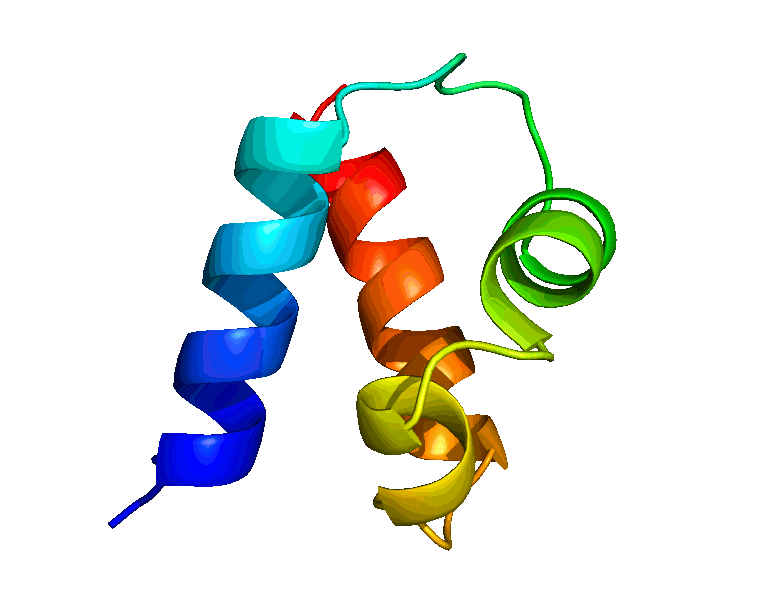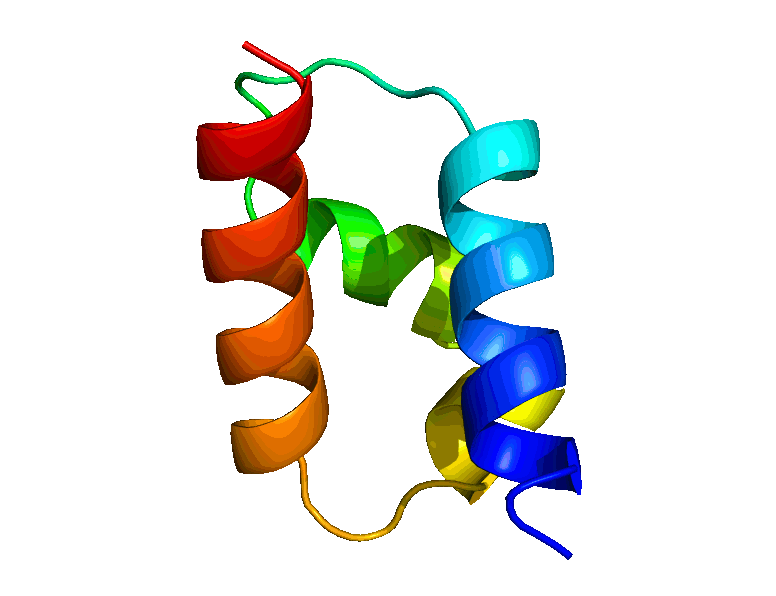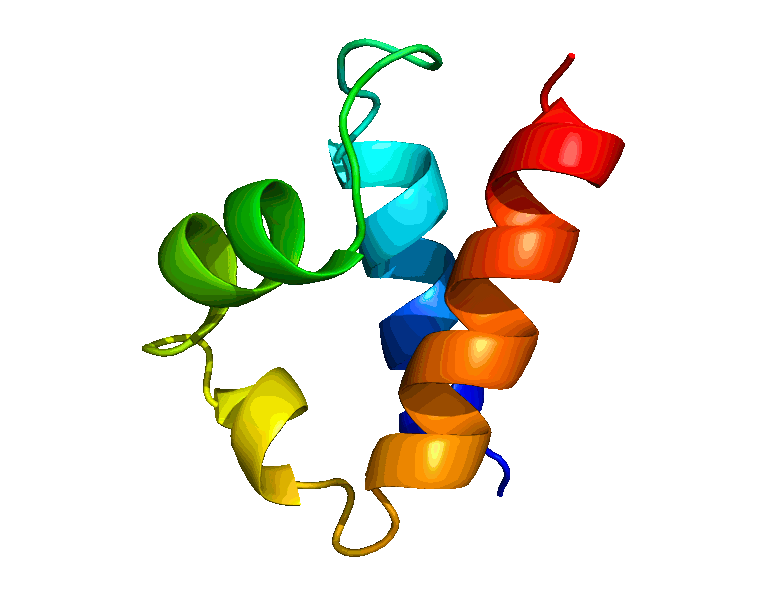
We have developed a suite of novel polynomial-time algorithms, called RDC-ANALYTIC [1, 3, 4], for high-resolution protein backbone structure determination from a minimal amount of residual dipolar coupling (RDC) and residual chemical shift anisotropy (RCSA) data. The core of RDC-ANALYTIC suite is based on exploiting the mathematical interplay between the sphero-conic representations of RDC and RCSA, and protein kinematics, to derive quartic equations, which can be solved analytically to obtain closed-form solutions to protein backbone dihedral angles and peptide plane orientations. Our algorithms led to the development of a new framework, RDC-PANDA [4], for for high-resolution protein structure determination, which was used prospectively to solve the solution structure of the FF Domain 2 of human transcription elongation factor CA150 (FF2) (PDB id: 2KIQ).

Recently, we have developed a suite of algorithms, called POOl [2], for computing high-quality loop conformations using only a minimal amount of RDC data measured in one alignment medium. POOL can compute highly-accurate ensembles of loop conformations even in the presence of a moderate level of dynamics. This work was featured as the Cover Article [2] in the journal PROTEINS: Structure, Function, and Bioinformatics.
References
- Extracting Structural Information from Residual Chemical Shift Anisotropy: Analytic Solutions for Peptide Plane Orientations and Applications to Determine Protein Structure. C. Tripathy, A. K. Yan, P. Zhou and B. R. Donald. The 17th Annual International Conference on Research in Computational Molecular Biology (RECOMB), Beijing, China, 2013. In Press.
- Protein Loop Closure using Orientational Restraints from NMR Data. C. Tripathy, J. Zeng, P. Zhou, and B. R. Donald. Proteins: Structure, Function, and Bioinformatics, 80(2):433-453, 2012. (Cover Article)
- Algorithms and Analytic Solutions using Sparse Residual Dipolar Couplings for High-Resolution Automated Protein Backbone Structure Determination by NMR. A. Yershova,* C. Tripathy,* P. Zhou, and B. R. Donald. In proceedings of the Ninth International Workshop on the Algorithmic Foundations of Robotics (WAFR, 2010), Springer Tracts in Advanced Robotics, 68:355-372, 2011. *These authors contributed equally to the work.
- High-Resolution Protein Structure Determination Starting with a Global Fold Calculated from Exact Solutions to the RDC Equations. J. Zeng, J. Boyles, C. Tripathy, L. Wang, A. Yan, P. Zhou, and B. R. Donald. Journal of Biomolecular NMR, 45(3):265-281, 2009.