

Nanjiang Shu

My current research interests are: (1) prediction of zinc-binding sites in proteins from sequences, (2) remote homology detection, and (3) prediction of secondary structures and shape strings (strings of labelled dihedral angles) of proteins.
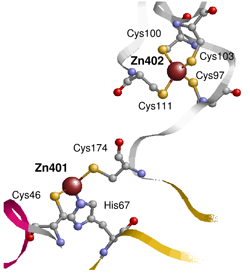
Motivated by the abundance, importance and unique functionality of zinc, both biologically and physiologically, we have developed an improved method for the prediction of zinc-binding sites (which contain 3 or 4 residues binding to a single zinc atom) in proteins from their amino acid sequences. Our method predicts zinc-binding residues with 75% precision (86% for Cys and His only) at 50% recall (the number of predicted zinc-binding residues out of all zinc-binding residues) when tested on a non-redundant set of SCOP (Structural Classification of Proteins) containing 2727 domains, which is 10% higher in accuracy than the most recently published method (Passerini et al., 2006).
The accurate prediction of zinc-binding residues in sequences can be used directly to screen zinc-binding proteins in genomes. The predicted zinc-binding proteins can be used to complete the current metalloprotein or catalytic site database, e.g. MDB (Metalloprotein Database and Browser) and CSA (Catalytic Site Atlas). In addition, the accurate prediction of zinc-binding proteins might be used to select protein enzymes capable of catalyzing inorganic reactions and mediating the formation of crystals, which is important in material synthesis. Our first version of zinc-binding prediction algorithm predicts whether a residue (or protein) binds to zinc or not, based on the fact that biological zinc-binding sites always have 3 or 4 residues binding to the same zinc atom. The prediction of the whole zinc-binding group, i.e. exactly which 3 or 4 residues that bind to the same zinc atom, is more challenging. The prediction of the whole zinc-binding group will be of great help for the metalloprotein design, and it will facilitate the 3D structure prediction greatly since the 3D space of the structure of zinc-binding proteins will be restricted enormously if zinc-binding sites can be allocated.
Breakthrough in the large-scale sequencing technique has led to a surge in biological sequence information. In this post-genomic era, functional annotation of the massive accumulated sequence information using computational methods becomes essential and is acquiring increasing interest. On the other hand, in structure prediction, despite decades of study, the homology modelling is still the most successful method. Traditionally, people use pairwise sequence alignment to detect homology between proteins. However, for sequences related at < 25% sequence identity, pairwise sequence alignment is normally unable to tell the homology between proteins.
To detect the homology between proteins with no obvious sequence similarity is a challenge. To date, the most sensitive methods to detect remote homologues rely on evolutionary information derived from multiple sequence alignments and protein structure information. The former includes profile-based methods, e.g. PSI-BLAST and SW-PSSM, and HMM (Hidden Markov Model) based methods, e.g. SAM and HMMSTR. The latter includes sequence-structure threading methods that can detect more distantly related proteins, e.g. 3D-PSSM and SVM-I-sites. We are developing a remote homology detection method using profile-based segment matching. Our method compares nine-residue long sliding fragments of the target sequence to all such fragments in the database by a profile-profile score. The profile is generated by combining the sequence profile generated by PSI-BLAST and a structural profile generated by shape string blocks. Our method competes with the best remote homology prediction methods. Moreover, our prediction outputs a dot plot of matched fragments of each homology candidate, which is intuitive and suitable for further checking of individual cases.
Predicting the secondary structure of proteins is usually the first step in the ab initio 3D structure prediction. However, the on average 45% so called random coils in the secondary structure expression brings enormous difficulties in the later stage of the 3D structure prediction, i.e., the packing of secondary structure elements into the native conformation. The protein backbone structure can also be represented by a set of dihedral angles due to the planarity of the peptide bond. By analyzing the high-resolution X-ray protein structures in PDB we found that these dihedral angles were generally clustered into eight regions in the Ramachandran plot. By applying a symbol to each of these clustered regions, the protein backbone structure can be represented by a one-dimensional string, called shape string, similar to the secondary structure representation of the protein structure. We are developing a method that combines the prediction of protein secondary structures and shape strings. At present, our method predicts protein secondary structures and shape strings at 83.7% Q3 accuracy and 71.2% Q8 (8-state shape strings) accuracy respectively, based on a non-redundant set of PDB culling at 30% sequence identity level, evaluated by a leave-one-out cross-validation. This set contains 4993 protein chains.
Moreover, our secondary structure prediction method predicts the total content of secondary structure in proteins quite accurately. The correlation coefficient for the predicted content of a-helices to the real content of a-helices is as high as 0.965, which is even higher than the experimental method such as Circular Dichroism which is both more time consuming and expensive. The shape string is as compact as the secondary structure expression of the protein backbone structure but carries more information in the loop regions; hence it can be used for fast searching of similar structures in the database.
References
- Shu, N., T. Zhou, et al. (2008). "Prediction of zinc-binding sites in proteins from sequence." Bioinformatics 24(6): 775-782.
- Shu, N., S. Hovmöller, et al. "Describing and Comparing Protein Structures.", Current Protein & Peptide Science, in press.